








How building automation impacts health, efficiency, and indoor air quality in hospitals
How does building automation ensure hospitals are safe, secure, comfortable environments for everyone?




Accountability is an obligation to understand and justify results. For built environment professionals and facility executives, accountability means being answerable for the significant influence of complex mechanical and electrical systems on business and mission-oriented key performance indicators. These professionals can be accountable to organizations, tenants, patients, students, shareholders, and consumers. In the articles Accountable operational technology and RC-WebView: The accountable BUI, we considered the important role facility automation and operational technology (OT) systems play in built environment accountability. In some heavily regulated industries, built environment professionals and facility executives are also accountable to the general public through stringent legislation and regulation.
One example of regulation that has worldwide influence is the Code of Federal Regulations (CFR) Title 21, which prescribes rules for the U.S. Food and Drug Administration (FDA). Part 11 establishes the requirements for electronic records and signatures in processes and facilities regulated by Title 21. Designed to govern technology systems that manage information in GxP1 processes, Title 21 has significant implications for pharmaceutical, medical device, biologic, biotech, contract research, and other regulated industries. Reliable Controls Authorized Dealers with RC-WebView® 3.13 are equipped to provide simple, flexible, and sustainable built environment accountability in these environments.
Title 21 regulates food and drugs manufactured or consumed in the United States under the jurisdiction of the FDA, the Drug Enforcement Administration, and the Office of National Drug Control Policy (Microsoft 2020). Although many countries have their own regulations and regulatory authorities for good practices in these fields, the size of the US market for food and pharmaceutical consumables makes Title 21 a common validation standard worldwide, particularly for manufacturing and logistics organizations.
Title 21 CFR Part 11 prescribes assurance that electronic records in regulated industry processes are accurate and available. This assurance is provided through the enforcement of improved cybersecurity, change control, electronic signatures, audit trails, and digital validation. IT and OT systems must have technological and procedural controls to protect data and ensure all records are authentic, incorruptible, and (where applicable) confidential (Ofni Systems n.d.). Specifically, the regulations “set forth the criteria under which the agency considers electronic records, [and] electronic signatures…to be trustworthy, [and] reliable” and state that digital records must be “readily available for, and subject to, FDA inspection” (Electronic Code of Federal Regulations [e-CFR] 2020).
Title 21 CFR Part 11 provides guidance for maintaining computer systems, including hardware and software, controls, and documentation in all regulated processes. All computer systems that store data that will be reported to the FDA or used to make quality decisions must be compliant. In laboratory situations, this includes records that prove quality, safety, strength, efficacy, or purity. In clinical environments, this includes data reported in clinical trials that determine quality, safety, or efficacy. In manufacturing environments, this includes decisions related to product quality. Critical to many of these metrics is indoor environmental control and monitoring during testing, manufacturing, and storage.
Title 21 CFR Part 11 provides the following definitions of fundamental concepts in electronic record-keeping and validation (e-CFR 2020):
For closed systems, where authenticated operators are accountable for the manipulation and management of data, procedures to ensure the authenticity, integrity, and confidentiality of electronic records are mandated when those records are created, modified, maintained, or transmitted. These controls apply to an automation system that monitors or controls a validated process or environment. Below are a few of the procedures designed to prevent claims by those accountable for electronic records that data is inaccurate or counterfeit (e-CFR 2020):
Open systems must follow the same requirements “from the point of their creation to the point of their receipt…and additional measures such as document encryption and use of appropriate digital signature standards to ensure…record authenticity, integrity, and confidentiality.”7
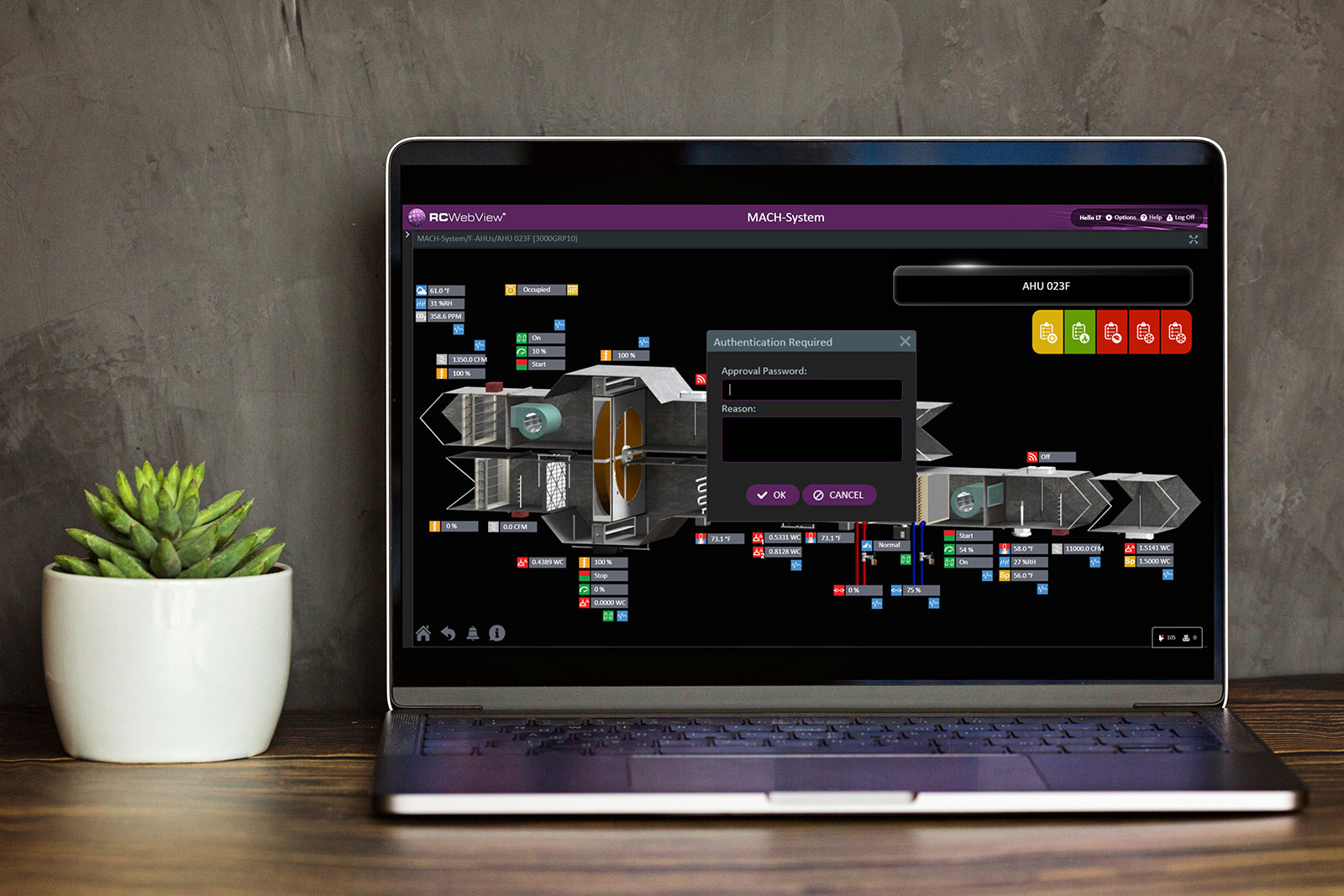
Electronic signatures are fundamental to providing accountability for electronic records. Changes to electronic records must be signed by, or digitally tied to, a specific person who is answerable for results. One of the means for providing this assurance is the use of “at least two distinct identification components such as an identification code and password.”8 This provision requires that a person authorized to manipulate digital records must have two distinct authenticators, perhaps one to access the system and another to change data. Although specific authentication must be provided for individual changes, the regulation allows for multiple changes during a single continuous period of controlled system access.
Finally, the regulation provides guidance for control of authenticators like passwords, including password maintenance policies, periodic testing, and collaborative user management.
The FDA does not certify IT/OT products or components as approved or compliant with Title 21 CFR Part 11; instead, the regulation provides prescriptive performance guidelines. IT/OT systems in FDA-regulated environments must be validated. Validation is a process, often performed by specialized third-party consultants, to confirm functional performance of an organization-level process or system. The validation process documents the operations the system performs, the system configuration required to perform correctly, and the testing that demonstrates system performance according to the defined specifications. Validation is performed for an entire process and typically includes the following components at a minimum:
RC-WebView can be a component of a Title 21 CFR Part 11−validated system for FDA-regulated processes and indoor environment monitoring or control. The intrinsic features of RC-WebView 3.13 described in RC-WebView: The accountable BUI make it appropriate for operating according to the regulation:
Validation requires close coordination with organizations, operations, and design teams as well as the validation consultant. Prior to the validation process, Reliable Controls Authorized Dealers will likely be called on to provide design documentation that describes how the system is designed and operational documentation that describes how it should be used according to the requirements of Title 21 CFR Part 11. There will likely also be requirements for operator training. Organizations need to articulate policies and procedures that provide process-level documentation and external security as well as policies for internal security, change control, and training. Many validation service providers offer helpful checklists that simplify design process and validation preparation.9
The RC-WebView Software Manual and online help provide guidance for the implementation of the intrinsic feature set expressly designed to satisfy the requirements of Title 21 CFR Part 11. There are a few simple but important additional design considerations.
The regulation is focused on accountability for electronic data. A significant portion of this answerability is provided by limiting who can make changes and keeping an accurate record of all changes, which can be done in a Reliable Controls system using RC-WebView. To ensure accountability, once deployed, only RC-WebView should be used to interact with the system. This is particularly important in validated systems. Once the validation process has begun and especially after complete, building operators should not deploy RC-Studio® as it could be used to make unaccountable changes.
Similarly, RC-Archive® should be provisioned using a custom installation without database utilities, as this feature set can also be used to modify electronic records without accountability.
Another consideration for validated systems is that using more smaller systems is more sustainable than using a larger validated enterprise. If any changes are made to a system, it is likely that the system would have to be revalidated. In a smaller segmented system, even simple changes that mandate revalidation have a lesser impact than if an entire enterprise encompassing several systems, users, and facilities has to be revalidated.
In industries regulated by the FDA, including pharmaceutical, medical device, biologic, biotech, and contract research, compliance with Title 21 CFR Part 11 is not optional or a value add; it is mandatory to daily operation. Before the COVID-19 pandemic, pharmaceutical spending in the United States was expected to grow to US$420 USD billion by 2023 (O’Brien 2019). Seventy percent of popular brand-name pharmaceuticals in the United States are imported (Gabriel Levitt 2017). This makes validation of OT systems in even this one regulated industry a global business opportunity. The next installment of Insight will discuss some ways we can bring our benefits to other FDA-regulated industries.
Built environment professionals and facility executives in regulated industries are accountable for seemingly countless key performance indicators. They have more than enough on their minds and endless demands on their time. RC-WebView 3.13 and Reliable Controls Authorized Dealers are an ideal combination to ease their burden with a simple, flexible, and accountable BUI for operational technology systems. By empowering these professionals with assurance that their electronic data is accurate and available, we can help them be better by design.
1 GxP guidelines established by the FDA regulate good practices. The “x” represents the variable disciplines in the pharmaceutical industry, including good manufacturing practice (GMP), good laboratory practice (GLP), good documentation practice (GDP), and good clinical practice (GCP).
2 21 CFR Part 11.10.d
3 21 CFR Part 11.10.g
4 21 CFR Part 11.10.e
5 21 CFR Part 11.10.c
6 21 CFR Part 11.10.a
7 21 CFR Part 11.11
8 21 CFR Part 11.200
9 An internet search for “Title 21 CFR Part 11 checklist” will return several examples of what to expect in a validation process.
Levitt, Gabriel. “How Can I Determine Where a Drug is Manufactured?” PharmacyChecker.com, October 15, 2017.
https://www.pharmacychecker.com/askpc/how-can-i-determine-where-a-drug-is-manufactured/#!.
Microsoft. Food and Drug Administration CFR Title 21 Part 11. March 23, 2020.
O’Brien, Jack. “Pharmaceutical Spending to Top $370B in 2019.” HealthLeaders, May 28, 2019.
https://www.healthleadersmedia.com/finance/pharmaceutical-spending-top-370b-2019.
Electronic Code of Federal Regulations (e-CFR). Title 21: Food and Drugs Part 11—Electronic Records; Electronic Signatures (2020). Code of Federal Regulations, National Archives and Records Administration (United States). Washington: Office of the Federal Register.
Ofni Systems. Introduction to 21 CFR Part 11 (n.d.).
http://www.ofnisystems.com/information/resources/introduction-to-21-cfr-11/.
ID 31828393 © Photographerlondon | Dreamstime.com
ID 161029793 © Alinabuzunova | Dreamstime.com

How does building automation ensure hospitals are safe, secure, comfortable environments for everyone?

Learn when and why MS/TP communication is helpful in building automation systems.

From the moment you park your car at a hockey arena, your comfort and safety are enhanced through a building automation system. Here’s how.

Building automation systems can make buildings truly intelligent, reducing their carbon footprint and saving money. Learn how.

Ever wonder how a building automation system ensures safety and accuracy in lab work?

Museums—and their building automation systems—play an important role in preserving the preserved. Learn how.

When you see a doctor or personal trainer, you benefit from their knowledge and experience. In the same way, a building automation system should provide all the tools you need to maintain your buildings in optimal health.

What are you doing to mitigate the transmission of viruses indoors? Learn what measures you can take and how your smart building provides more information and control for IAQ concerns.